¡Hola de nuevo! En primer lugar, quería desearos unas felices fiestas, ya que estamos casi terminando el año. Vamos a hablar ahora del síndrome de Hurler.
Este es una enfermedad rara con una baja incidencia, que está aproximadamente en un caso por cada 175000 nacimientos. Esta enfermedad es una enfermedad hereditaria.
Quizá habéis oído hablar alguna vez de las mucopolisacaridosis. Pues el síndrome de Hurler es la forma más grave de la mucopolisacaridosis tipo 1. Esta es una enfermedad que va a afectar al metabolismo. Lo que ocurre es que está ausente una enzima que es la alfa-L-iduronidasa lisosómica, que es la encargada de degradar los glucosaminoglicanos o mucopolisacáridos. Entonces, lo que sucede es que se van a acumular y van a producir destrucción en algunos órganos.
Una vez dicho en qué consiste la enfermedad, lo que más os rondará la cabeza en estos momentos es saber cómo se puede detectar que se está produciendo esto en el cuerpo.
El síndrome de Hurler abarca muchos signos y síntomas, y se pueden observar a nivel físico, a nivel intelectual, a nivel de los distintos órganos...
lunes, 29 de diciembre de 2014
Síndrome de Goodpasture
¡Buenos días a todos! Hoy hablaremos entre otros, del síndrome de Goodpasture. De nuevo estamos ante una enfermedad rara que tiene una incidencia estimada en 0,1 casos por cada millón de habitantes, una de las enfermedades con menos incidencias de las que hemos visto en el blog. Además, es importante saber que afecta en mayor proporción a hombres que a mujeres.
Siendo tan poco frecuente, quizás no habréis oído hablar de ella ni de sus casos ni de sus síntomas, y por ello, los vamos a explicar aquí.
Esta enfermedad es del grupo de las enfermedades autoinmunitarias, es decir, que el propio sistema inmunitario de la persona va a ser atacado a sí mismo por equivocación, destruyendo tejidos corporales que se encontraban en perfecto estado. Concretamente, esta enfermedad va a comprometer a los riñones y a los pulmones, produciéndose insuficiencia renal que evolucionará de manera rápida y enfermedad pulmonar.
Siendo tan poco frecuente, quizás no habréis oído hablar de ella ni de sus casos ni de sus síntomas, y por ello, los vamos a explicar aquí.
Esta enfermedad es del grupo de las enfermedades autoinmunitarias, es decir, que el propio sistema inmunitario de la persona va a ser atacado a sí mismo por equivocación, destruyendo tejidos corporales que se encontraban en perfecto estado. Concretamente, esta enfermedad va a comprometer a los riñones y a los pulmones, produciéndose insuficiencia renal que evolucionará de manera rápida y enfermedad pulmonar.
 |
| Fuente: www.epainassist.com |
Elefantiasis
Se trata de un síndrome caracterizado por el enorme aumento de ciertas partes del cuerpo, especialmente de las extremidades inferiores y de los órganos genitales externos. Puede producirse por diversas enfermedades inflamatorias persistentes, producida por diversas espiroquetas, treponemas y parásitos.

Se debe a la obstrucción de los vasos linfáticos y es por esto que da como resultado inflamaciones severas y de parásitos sanguíneos. También deriva en la malformación ósea, la cual va deformando el cuerpo hasta un cierto límite.
Hay algunas opciones para el tratamiento de la elefantiasis, incluyendo antibióticos, regímenes estrictos de limpieza y el ejercicio. Mantener la piel limpia y seca, incluso cuando la hinchazón es muy grande, resulta importante para evitar el desarrollo y la propagación de la infección.
Fuentes: http://elefantiasis.org/
Mielofibrosis
Se trata de un trastorno de la médula ósea en el que la médula se ve desplazada en gran medida por tejido fibroso. Así, la cicatrización de la médula ósea supone que esta médula no es capaz de producir el número suficiente de células sanguíneas por lo que se puede presentar anemia, problemas de sangrado y un riesgo grande de sufrir infecciones.
Para compensar esto, tanto el hígado como el bazo tratan de producir algunas de estas células sanguíneas lo que provoca la inflamación de tales órganos, denominada hematopoyesis extramedular.
La causa de la mielofibrosis es desconocida por el momento y se suele dar en personas de edad superior a los 50 años.
En el amplio abanico de síntomas hablaremos de la llenura abdominal (aumento del tamaño del bazo), dolor óseo, hematomas, sangrado fácil, fatiga, palidez y dificultad para respirar al hacer ejercicio.

Fuentes: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000531.htm
Para compensar esto, tanto el hígado como el bazo tratan de producir algunas de estas células sanguíneas lo que provoca la inflamación de tales órganos, denominada hematopoyesis extramedular.
La causa de la mielofibrosis es desconocida por el momento y se suele dar en personas de edad superior a los 50 años.
En el amplio abanico de síntomas hablaremos de la llenura abdominal (aumento del tamaño del bazo), dolor óseo, hematomas, sangrado fácil, fatiga, palidez y dificultad para respirar al hacer ejercicio.
Fuentes: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/000531.htm
Tricotilomanía
Es un tipo de trastorno de control impulsivo en el que encontramos la pérdida de cabello por ganas de halarlo y retorcerlo hasta que se desprende. Los afectados son incapaces de cesar este comportamiento incluso cuando su pelo se vuelve más delgado. Las causas no se comprenden con claridad y las mujeres tienen una probabilidad 4 veces mayor de verse afectadas que los hombres.
Los síntomas comienzan antes de los 17 años y el cabello se pierde de forma progresiva por parches redondos a lo largo del cuero cabelludo, lo que causa un efecto de apariencia desigual. También se puede dar el caso de que la persona se arranque otras áreas de cabello como las cejas, las pestañas o el vello corporal.
Entre los síntomas podemos destacar el bloqueo intestinal si las personas ingieren el cabello que arrancan, el cabello que vuelve a crecer en áreas descubiertas aparece en forma de cerdas, ciertos comportamientos de autoagresión y sensación de placer y gratificación tras halarse el pelo.
Al referirnos al tratamiento, la naltrexona y los inhibidores selectivos de la recaptación de la serotonina han demostrado ser eficaces para determinados síntomas.

Fuentes: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/001517.htm
Los síntomas comienzan antes de los 17 años y el cabello se pierde de forma progresiva por parches redondos a lo largo del cuero cabelludo, lo que causa un efecto de apariencia desigual. También se puede dar el caso de que la persona se arranque otras áreas de cabello como las cejas, las pestañas o el vello corporal.
Entre los síntomas podemos destacar el bloqueo intestinal si las personas ingieren el cabello que arrancan, el cabello que vuelve a crecer en áreas descubiertas aparece en forma de cerdas, ciertos comportamientos de autoagresión y sensación de placer y gratificación tras halarse el pelo.
Al referirnos al tratamiento, la naltrexona y los inhibidores selectivos de la recaptación de la serotonina han demostrado ser eficaces para determinados síntomas.
Fuentes: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/001517.htm
Glosodinia o síndrome de la boca ardiente
Es una enfermedad muy poco frecuente de origen desconocido que se caracteriza porque los pacientes presentas sensaciones muy dolorosas de ardor en la cavidad bucal. Los afectados refieren una sensación de quemazón constante en la boca pero al explorar la mucosa y al llevar a cabo las pruebas analíticas no se detectan anormalidades.
En cuanto a los síntomas más frecuentes de la patología cabe destacar la quemazón, lengua reseca o entumecida además del picor y hormigueo, encías doloridas e hipersensibles, paladar abrasivo, labios cortados, sensación de sabor metálico y sequedad en la boca sin aparente hiposalivación.
En la actualidad, existe una importante controversia para determinar si se trata de un trastorno de origen fisiológico o si, por el contrario, constituye la manifestación de alteraciones psicosomáticas.

Fuentes: https://www.propdental.es/blog/odontologia/tratamiento-sindrome-de-la-boca-ardiente/
En cuanto a los síntomas más frecuentes de la patología cabe destacar la quemazón, lengua reseca o entumecida además del picor y hormigueo, encías doloridas e hipersensibles, paladar abrasivo, labios cortados, sensación de sabor metálico y sequedad en la boca sin aparente hiposalivación.
En la actualidad, existe una importante controversia para determinar si se trata de un trastorno de origen fisiológico o si, por el contrario, constituye la manifestación de alteraciones psicosomáticas.
Fuentes: https://www.propdental.es/blog/odontologia/tratamiento-sindrome-de-la-boca-ardiente/
viernes, 26 de diciembre de 2014
Síndrome de argiria
Es una patología poco conocida que se caracteriza por el cambio de color de la piel, otros órganos y tejidos de su color habitual a un tono grisáceo o gris azulado, siendo especialmente acusado este cambio en las zonas expuestas al sol. Tiene por causa la ingesta prolongada de plata metálica o de alguna de sus sales. Existen casos de argiria localizada o generalizada.
La plata se puede depositar en el organismo por exposición industrial o también a través de ciertos medicamentos que contienen sales de plata por lo que la mayoría de los casos los encontraremos en trabajadores de la minería y fabricación de aleaciones metálicas.
El tratamiento con preparaciones despigmentantes no es demasiado eficaz si bien es cierto que las investigaciones apuntan a que los tratamientos con hidroquinona pueden reducir el número de gránulos de plata en la dermis superior y alrededor de las glándulas sudoríparas, además de disminuír el número de melanocitos.

La plata se puede depositar en el organismo por exposición industrial o también a través de ciertos medicamentos que contienen sales de plata por lo que la mayoría de los casos los encontraremos en trabajadores de la minería y fabricación de aleaciones metálicas.
El tratamiento con preparaciones despigmentantes no es demasiado eficaz si bien es cierto que las investigaciones apuntan a que los tratamientos con hidroquinona pueden reducir el número de gránulos de plata en la dermis superior y alrededor de las glándulas sudoríparas, además de disminuír el número de melanocitos.
Epidermodisplasia verruciforme
Es un raro trastorno hereditario de la piel asociado a un alto riesgo de carcinoma. Se caracteriza por una gran susceptibilidad al virus del papiloma humano (VPH) de forma que el no control de la infección trae consigo la aparición de máculas y pápulas, especialmente en manos y pies.
La causa es genética y se debe a una mutación que inactiva los genes TMC6 o TMC8, que están situados uno junto al otro en el cromosoma 17. Estos genes desempeñan una labor en la distribución del zinc en el núcleo celular.
El paciente presenta unas ligeras escamas, máculas de color rojo en la cara, cuello y cuerpo, verrugas, lesiones de queratosis seborreica y pápulas de color rosa en extremidades.
Para su tratamiento se sugiere la administración de acitretina además de la actuación con interferones.

La causa es genética y se debe a una mutación que inactiva los genes TMC6 o TMC8, que están situados uno junto al otro en el cromosoma 17. Estos genes desempeñan una labor en la distribución del zinc en el núcleo celular.
El paciente presenta unas ligeras escamas, máculas de color rojo en la cara, cuello y cuerpo, verrugas, lesiones de queratosis seborreica y pápulas de color rosa en extremidades.
Para su tratamiento se sugiere la administración de acitretina además de la actuación con interferones.
Degeneración hepatolenticular
Se trata de un trastorno hereditario en el cual el organismo absorbe y conserva demasiado cobre de manera que éste se depositará en el hígado, cerebro, riñones y ojos. Los depósitos de cobre causan un fuerte daño tisular, muerte del tejido y cicatrización, lo cual hace que los órganos afectados dejen de funcionar correctamente.
¿Cómo determinar que uno está afectado? El examen de los ojos con lámpara de hendidura mostrará un movimiento ocular limitado además de un anillo de color marrón o rojizo alrededor del iris. Por su parte, el examen físico puede determinar daños en el Sistema Nervioso Central reflejados en la pérdida de coordinación o temblores musculares. En el ámbito del laboratorio, la bilirrubina elevada o niveles bajos de albúmina son también característicos.
El tratamiento consistirá en la reducción de cobre de los tejidos y ésto se hace a través de un proceso denominado quelación, en donde ciertos medicamentos se fijan al cobre y ayudan a eliminarlo a través de los riñones o de los intestinos.

¿Cómo determinar que uno está afectado? El examen de los ojos con lámpara de hendidura mostrará un movimiento ocular limitado además de un anillo de color marrón o rojizo alrededor del iris. Por su parte, el examen físico puede determinar daños en el Sistema Nervioso Central reflejados en la pérdida de coordinación o temblores musculares. En el ámbito del laboratorio, la bilirrubina elevada o niveles bajos de albúmina son también característicos.
El tratamiento consistirá en la reducción de cobre de los tejidos y ésto se hace a través de un proceso denominado quelación, en donde ciertos medicamentos se fijan al cobre y ayudan a eliminarlo a través de los riñones o de los intestinos.
Síndrome de Okamoto
Es una patalogía rarísima de la que sólo se ha tenido conocimiento de 6 casos en la historia, 4 de los cuales siguen con vida. Se compone de una serie de anomalías congénitas entre las que destacan el paladar hendido, la estenosis de la unión uteropélvica con hidronefrosis, problemas cardíacos, hipotonía generalizada, dificultad para el crecimiento y retraso del desarrollo.
Los rasgos faciales son muy característicos e incluyen microcefalia, hipoplasia de media cara, ojos prominentes, largas pestañas, lóbulo de la oreja largo, puente nasal deprimido, apariencia de boca abierta, labio inferior grande y comisuras de la boca hacia abajo.
Los afectados no pueden caminar sin ayuda. Debido a la hipotonía tampoco son capaces de masticar ni de hablar y cabe destacar la presencia de ciertos rasgos de autismo sin llegar a ser autistas. No existe cura y el desconocimiento es bastante grande debido a la escasez de afectados.

Fuentes: http://www.atencionprimariasalud.es/documentacion-otros-medios/vivir-con-una-enfermedad-rara-sindrome-de-okamoto
Los rasgos faciales son muy característicos e incluyen microcefalia, hipoplasia de media cara, ojos prominentes, largas pestañas, lóbulo de la oreja largo, puente nasal deprimido, apariencia de boca abierta, labio inferior grande y comisuras de la boca hacia abajo.
Los afectados no pueden caminar sin ayuda. Debido a la hipotonía tampoco son capaces de masticar ni de hablar y cabe destacar la presencia de ciertos rasgos de autismo sin llegar a ser autistas. No existe cura y el desconocimiento es bastante grande debido a la escasez de afectados.
Fuentes: http://www.atencionprimariasalud.es/documentacion-otros-medios/vivir-con-una-enfermedad-rara-sindrome-de-okamoto
La triste realidad de Tay-Sachs
La enfermedad de Tay-Sachs es un trastorno hereditario extremadamente poco común que se debe a un exceso de acumulación de una sustancia granosa en los tejidos y en las células nerviosas, acumulación que destruye de manera progresiva las neuronas y causa graves problemas físicos y mentales.
Los recién nacidos con esta patología parecen desarrollarse con normalidad durante los primeros meses de vida pero, a medida que las células se agrandan con el material granoso, las capacidades mentales y físicas se van deteriorando. El niño pierde la vista, la audición, no puede tragar, los músculos comienzan a atrofiarse y es visible la parálisis. Desgraciadamente, y aún con los mejores cuidados, los niños con Tay-Sachs suelen morir antes de los 4 años.
No existe cura, las medicinas adecuadas y una buena nutrición puedes ayudar con ciertos síntomas y cabe destacar que gran parte de los niños necesitan sonda para alimentarse.

Fuente: http://www.nlm.nih.gov/medlineplus/spanish/taysachsdisease.html
Los recién nacidos con esta patología parecen desarrollarse con normalidad durante los primeros meses de vida pero, a medida que las células se agrandan con el material granoso, las capacidades mentales y físicas se van deteriorando. El niño pierde la vista, la audición, no puede tragar, los músculos comienzan a atrofiarse y es visible la parálisis. Desgraciadamente, y aún con los mejores cuidados, los niños con Tay-Sachs suelen morir antes de los 4 años.
No existe cura, las medicinas adecuadas y una buena nutrición puedes ayudar con ciertos síntomas y cabe destacar que gran parte de los niños necesitan sonda para alimentarse.
Fuente: http://www.nlm.nih.gov/medlineplus/spanish/taysachsdisease.html
lunes, 22 de diciembre de 2014
Síndrome de Sanfilippo
Se trata de un trastorno hereditario del metabolismo por el cual el organismo no es capaz de descomponer apropiadamente cadenas largas de moléculas de azúcar denominadas glucosaminoglicanos.
Se debe a una carencia en la cantidad de enzimas necesarias para descomponer la cadena de azúcares de heparán sulfato. Distinguimos 4 formas distintas del síndrome de Sanfilippo en función de la enzima que se encuentre afectada:
- Tipo A: es la forma más grave y los afectados carecen o poseen una forma alterada de la enzima heparán N-sulfatasa.
Se debe a una carencia en la cantidad de enzimas necesarias para descomponer la cadena de azúcares de heparán sulfato. Distinguimos 4 formas distintas del síndrome de Sanfilippo en función de la enzima que se encuentre afectada:
- Tipo A: es la forma más grave y los afectados carecen o poseen una forma alterada de la enzima heparán N-sulfatasa.
Síndrome de Lemierre
También se denomina sepsis post angina y se trata de una complicación muy poco frecuente de la amigdalitis por una sepsis grave. Se caracteriza por la aparición de tromboflebitis en la vena yugular interna.
Suele estar causada por una bacteria denominada fusobacterium necrophorum que es un gérmen anaerobio habitual en la flora de la cavidad bucal.
La enfermedad fue descrita por primera vez a través de la figura del médico André Lemierre que, en 1936, publicó un artículo en el que describía una serie de 20 casos, entre los cuales hubo una gran mortalidad.
Suele estar causada por una bacteria denominada fusobacterium necrophorum que es un gérmen anaerobio habitual en la flora de la cavidad bucal.
La enfermedad fue descrita por primera vez a través de la figura del médico André Lemierre que, en 1936, publicó un artículo en el que describía una serie de 20 casos, entre los cuales hubo una gran mortalidad.
Pólipos de colon
Nos referimos a una serie de prominencias en el interior del intestino grueso que aparecen en la mucosa que recubre el interior del colon. En ocasiones pueden llegar a ser muy numerosas y, entonces, hablaremos de un grupo de enfermedades denominadas poliposis.
La importancia de estos pólipos estriba en el hecho de que en su composición puede haber células con cambios precancerosos, son los llamados pólipos adenomatosos. Estos pólipos pueden llegar a degenerar todavía más con el paso de los años hasta dar lugar a un cáncer de colon. Su extirpación cuando aún miden pocos milímetros evitaría la aparición de ese hipotético cáncer.
Síndrome de Kabuki
Es una patalogía rara caracterizada por anomalías congénitas múltiples como rasgos faciales típicos, anomalías esqueléticas, discapacidad intelectual entre leve y moderada y déficit de crecimiento postnatal.
Los rasgos craneofaciales incluyen fisuras palpebrales alargadas, cejas arqueadas y espesas pero en su tercio lateral escasamente pobladas, orejas prominentes, labio leporino, paladar hendido, problemas dentales, etc. También son frecuentes los defectos congénitos del corazón, las anomalías renales y del conducto urinario y la pérdida de audición.
Los rasgos craneofaciales incluyen fisuras palpebrales alargadas, cejas arqueadas y espesas pero en su tercio lateral escasamente pobladas, orejas prominentes, labio leporino, paladar hendido, problemas dentales, etc. También son frecuentes los defectos congénitos del corazón, las anomalías renales y del conducto urinario y la pérdida de audición.
Esclerosis tuberosa
Se trata de una enfermedad de origen genético, consistente en el crecimiento de tumores benignos (hamartomas), y malformaciones en uno o varios órganos como pueden ser la piel, cerebro, riñones, corazón, ojos, pulmones, dientes, etc.
La afectación de la patología es muy variable ya que va desde pequeñas alteraciones cutáneas hasta un retraso mental importante y problemas en múltiples órganos.
La esclerosis tuberosa es hereditaria y se debe a la mutación presente en los genes TSC1 y TSC2, siendo ésta la causa de la mayoría de los casos de esta afección.
La afectación de la patología es muy variable ya que va desde pequeñas alteraciones cutáneas hasta un retraso mental importante y problemas en múltiples órganos.
La esclerosis tuberosa es hereditaria y se debe a la mutación presente en los genes TSC1 y TSC2, siendo ésta la causa de la mayoría de los casos de esta afección.
Trimetilaminuria o Síndrome del olor a pescado
Es una enfermedad muy poco frecuente que hace que la persona que la sufre desprenda un fuerte olor a pescado podrido a través del sudor, del aliento, de la orina y, en el caso de las mujeres, también a través de las secreciones vaginales.
Su causa se debe a la falta de asimilación de uno de los componentes de los alimentos tales como las legumbres, los guisantes, la yema de huevo, los pescados de agua salada o el marisco. Todos ellos tienen presente la trimetilamina en su composición que, unido al fallo metabólico del paciente, provoca su oxidación que tiene lugar en el hígado.
Su causa se debe a la falta de asimilación de uno de los componentes de los alimentos tales como las legumbres, los guisantes, la yema de huevo, los pescados de agua salada o el marisco. Todos ellos tienen presente la trimetilamina en su composición que, unido al fallo metabólico del paciente, provoca su oxidación que tiene lugar en el hígado.
sábado, 20 de diciembre de 2014
Pénfigo familiar benigno
Es una patología caracterizada por la presencia de fisuras en la piel, principalmente en las axilas y en los pliegues inguinales y perineales (vulva y escroto). Las lesiones en la piel aparecen durante la adolescencia o, de manera más frecuente, sobre los 30-40 años de edad.
Tiene una transmisión de tipo autosómico dominante con penetrancia incompleta y decimos que viene causado por una serie de mutaciones en el gen ATP2C1, el cual codifica para una bomba de calcio. provocando la enfermedad al impedir la adhesión de los queratinocitos epidérmicos.
Tiene una transmisión de tipo autosómico dominante con penetrancia incompleta y decimos que viene causado por una serie de mutaciones en el gen ATP2C1, el cual codifica para una bomba de calcio. provocando la enfermedad al impedir la adhesión de los queratinocitos epidérmicos.
Neuromielitis óptica
Es una enfermedad desmielinizante del Sistema Nervioso Central caracterizada por la afección preferentemente del nervio óptico y de la médula espinal. La patología es 9 veces más frecuente es mujeres que en hombres y la edad media para su aparición se sitúa en torno a los 39 años.
Es una enfermedad que presenta un curso caracterizado por brotes, generalmente con recuperación incompleta y una discapacidad que va incrementando en relación a la gravedad de dichos brotes. Más de la mitad de los afectados acaban ciegos de uno o ambos ojos o bien necesitan ayuda para caminar una vez que finalizan los 5 primeros años con la enfermedad. Fue considerada una variante de esclerosis múltiple pero asociada a otro pronóstico y a otra respuesta terapéutica.
Es una enfermedad que presenta un curso caracterizado por brotes, generalmente con recuperación incompleta y una discapacidad que va incrementando en relación a la gravedad de dichos brotes. Más de la mitad de los afectados acaban ciegos de uno o ambos ojos o bien necesitan ayuda para caminar una vez que finalizan los 5 primeros años con la enfermedad. Fue considerada una variante de esclerosis múltiple pero asociada a otro pronóstico y a otra respuesta terapéutica.
Osteocondritis de tobillo
Se trata de una enfermedad grave, poco común, pero que afecta a una articulación de enorme importancia como es el tobillo. Cuando hablamos de esta patología cabe destacar que nos referimos a la osteocondritis del astrálago, uno de los tres huesos que conforman el tobillo junto con la tibia y el peroné. La parte superior de este hueso está completamente cubierta por cartílago y se articula con la tibia, por lo que se trata de un hueso de gran importancia motora.
La osteocondritis es literalmente una llaga que aparece en el cartílago. Una porción de este cartílago experimenta un proceso de muerte celular en la parte en la que se produce el contacto con el hueso.
Visualmente existe un reblandecimiento del cartílago que puede derivar en un desprendimiento, quedando el hueso subyacente al descubierto y pudiendo generar lo que se conoce como un cuerpo libre.
La osteocondritis es literalmente una llaga que aparece en el cartílago. Una porción de este cartílago experimenta un proceso de muerte celular en la parte en la que se produce el contacto con el hueso.
Visualmente existe un reblandecimiento del cartílago que puede derivar en un desprendimiento, quedando el hueso subyacente al descubierto y pudiendo generar lo que se conoce como un cuerpo libre.
jueves, 18 de diciembre de 2014
Enfermedad de Krabbe
¡¡Buenas tardes un día más!! ¿Qué os están pareciendo nuestras entradas? Si os interesan, hoy os dejo otra que habla acerca de la enfermedad de Krabbe. Quizá muchos de vosotros no habréis oído hablar de ella, y por eso yo os voy a explicar un poco de qué se trata.
La enfermedad de Krabbe es una enfermedad que afecta al sistema nervioso de la siguiente manera: existe una alteración en el gen GALC, lo que hace que exista deficiencia de la galactocerebrosidasa (una enzima). Al ocurrir esto, van a acumularse galactocerebrósidos, y además, no habrá suficiente mielina. Esto provoca también que las neuronas y los nervios cerebrales se vean afectados, desencadenando todo esto en un trastorno neurológico y degenerativo.
Probablemente no habréis escuchado mucho sobre ella porque su incidencia es bastante baja. De nuevo, estamos ante una enfermedad rara en la que la incidencia se estima en un caso por cada 100000 nacimientos, y en países como Francia, incluso 1 caso por cada 150000 nacimientos.
La enfermedad de Krabbe es hereditaria, siguiendo un patrón autosómico recesivo, por lo que a veces es frecuente que exista más de un caso en la misma familia.
La enfermedad de Krabbe es una enfermedad que afecta al sistema nervioso de la siguiente manera: existe una alteración en el gen GALC, lo que hace que exista deficiencia de la galactocerebrosidasa (una enzima). Al ocurrir esto, van a acumularse galactocerebrósidos, y además, no habrá suficiente mielina. Esto provoca también que las neuronas y los nervios cerebrales se vean afectados, desencadenando todo esto en un trastorno neurológico y degenerativo.
Probablemente no habréis escuchado mucho sobre ella porque su incidencia es bastante baja. De nuevo, estamos ante una enfermedad rara en la que la incidencia se estima en un caso por cada 100000 nacimientos, y en países como Francia, incluso 1 caso por cada 150000 nacimientos.
La enfermedad de Krabbe es hereditaria, siguiendo un patrón autosómico recesivo, por lo que a veces es frecuente que exista más de un caso en la misma familia.
Síndrome de Korsakoff
Se define como un trastorno de tipo cerebral que se produce por la deficiencia de tiamina.
CAUSAS:
El daño cerebral que se produce, tiene lugar debido a la baja cantidad de vitamina B existente en el organismo.
La falta de vitamina B1 es característica de personas que sufren de problemas de alcoholismo.

CAUSAS:
El daño cerebral que se produce, tiene lugar debido a la baja cantidad de vitamina B existente en el organismo.
La falta de vitamina B1 es característica de personas que sufren de problemas de alcoholismo.
Tambíen puede tener lugar en personas cuya absorción de alimentos no se realiza correctamente.
SÍNTOMAS:
-Incapacidad para formar nuevos recuerdos.
-Pérdida de memoria.
-Inventar historias.
-Alucinaciones.
Tumor de Wilms
Esta última entrada estará centrada en la enfermedad del TUMOR DE WILMS, un tumor renal que se presenta en los niños.
Etiología
La causa es desconocida pero existen anomalías asociadas con esta enfermedad como la aniridia, problemas urinarios o la hemipertrofia. Se presenta en niños de entre tres a ocho años de edad y se sospecha que la etiología sea de carácter genético ya que es común entre hermanos y gemelos.
Síntomas
Los niños con tumor de Wilms tienen dolor e hinchazón en el abdomen. Sienten un malestar general acompañado de fiebre, inapetencia, náuseas y vómitos. Otros síntomas pueden ser el color anormal de la orina, el estreñimiento, la tensión arterial alta y el aumento del tamaño de un solo lado del cuerpo.

miércoles, 17 de diciembre de 2014
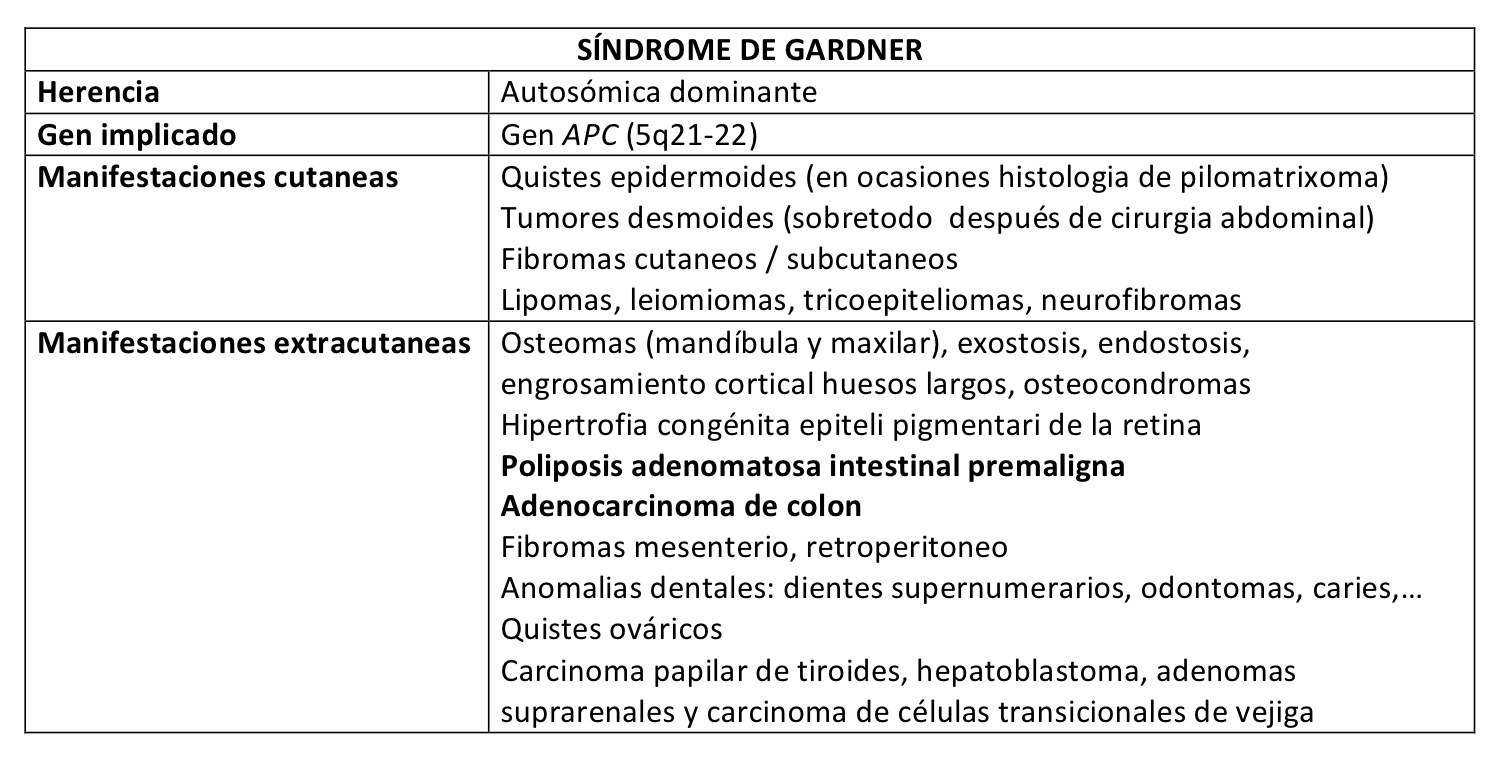
Síndrome de Gardner
Para hablar del síndrome de Gardner, tenemos que explicar antes el concepto de poliposis adenomatosa familiar. Esto es una enfermedad de tipo hereditaria, no frecuente, que se incluye dentro de los síndromes de poliposis intestinal, caracterizados por la aparición de pólipos de tipo adenomatoso.
El síndrome de Gadner, es la poliposis adenomatosa familiar atenuada, donde la cantidad de pólipos presentes es menos a 100 y manteniendo un tipo de herencia autosómica dominante.
Cabe destacar que es un factor de riesgo para que se produzca el desarrollo de un cáncer de tipo colorrectal, aunque también puede ser un factor de riesgo para la aparición de otro tipo de cánceres.
El síndrome de Gardner está caracterizado por los siguientes signos y síntomas:
El síndrome de Gadner, es la poliposis adenomatosa familiar atenuada, donde la cantidad de pólipos presentes es menos a 100 y manteniendo un tipo de herencia autosómica dominante.
Cabe destacar que es un factor de riesgo para que se produzca el desarrollo de un cáncer de tipo colorrectal, aunque también puede ser un factor de riesgo para la aparición de otro tipo de cánceres.
El síndrome de Gardner está caracterizado por los siguientes signos y síntomas:
Fibrodisplasia osificante progresiva (FOP): enfermedad del hombre estatua
¡¡Buenas tardes a todos nuestros lectores!! Vamos a dedicar hoy una de nuestras entradas a que conozcáis un poco más acerca de la fibrodisplasia osificante progresiva (FOP de aquí en adelante). Esta enfermedad consiste en la formación de hueso en músculos, ligamentos, tendones y tejidos blandos. No obstante, hay una serie de músculos que, generalmente, no suelen verse afectados, como son el corazón, el diafragma o la lengua. Con todo, una persona que tenga fibrodisplasia osificante progresiva tendrá los movimientos y la alimentación bastante limitados.
Supongo que os preguntaréis por qué se produce la FOP. Pues esto es debido a la activación de un gen mutado: el ACVR1, que estimula el crecimiento de tejido óseo incluso en zonas en las que no debía de haberlo, como es el caso.
Supongo que os preguntaréis por qué se produce la FOP. Pues esto es debido a la activación de un gen mutado: el ACVR1, que estimula el crecimiento de tejido óseo incluso en zonas en las que no debía de haberlo, como es el caso.
 |
| Fuente: www.lasenfermedades.org |
Tetralogía de Fallot
Buenas tardes! Hoy vamos a hablar de la TETRALOGÍA DE FALLOT. Una enfermedad congénita que afecta al corazón y a los vasos sanguíneos y que causa cianosis debido a una deficiencia de oxígeno en la sangre.
La tetralogía de Fallot incluye cuatro anormalidades cardíacas:
1. Comunicación entre los ventrículos
2. Estrechamiento de la arteria pulmonar (estenosis pulmonar)
3. Dextraposición de la aorta
4. Engrosamiento ventricular derecho

Síndrome de Alagille
Se define como una dolencia genética que afecta normalmente alhígado y al corazón, aunque también puede afectar a otros sistemas.
Sus síntomas comienza a notarse desde la infancia.
Estamos ante una enfermedad de tipo hereditario, que sigue un patrón de herencia autosómica dominante.
Las mutaciones en el gen JAG del cromosoma número 20 son las causantes de esta enfermedad.
SIGNOS Y SÍNTOMAS:
-Ictericia
-Manchas en los ojos
-Xantelasmas
-Alteraciones musculares
-Alteraciones faciales: hipertelorismo, mentón puntiagudo y frente ancha

Sus síntomas comienza a notarse desde la infancia.
Estamos ante una enfermedad de tipo hereditario, que sigue un patrón de herencia autosómica dominante.
Las mutaciones en el gen JAG del cromosoma número 20 son las causantes de esta enfermedad.
SIGNOS Y SÍNTOMAS:
-Ictericia
-Manchas en los ojos
-Xantelasmas
-Alteraciones musculares
-Alteraciones faciales: hipertelorismo, mentón puntiagudo y frente ancha
TRATAMIENTO:
No existe una cura para esta enfermedad. El tratamiento será sintomático y sus herramientas principales serán farmacológicas y la cirugía.
Síndrome de Adie
Es un trastorno neurológico que afecta a la pupila del ojo y al sistema nervioso autónomo.
Es causada debido a un daño en las fibras postganglionares de la inervación parasimpática del ojo, debido normalmente, a una infección viral o bacteriana, que provoca inflamación.
Se caracteriza por una pupila tónica dilatada.

Es causada debido a un daño en las fibras postganglionares de la inervación parasimpática del ojo, debido normalmente, a una infección viral o bacteriana, que provoca inflamación.
Se caracteriza por una pupila tónica dilatada.
martes, 16 de diciembre de 2014
Síndrome de Meige
Es una distonía focal que se caracteriza por la combinación de blefaroespasmo asociado con:
-Distonía laríngea.
-Faríngea
-Oromandibular
-Distonía bucolingual.
CAUSA:
Actualmente es de origen desconocido.
DIAGNÓSTICO:
Es frecuente que se produzcan errores de diagnóstico, pues no se poséen las pruebas o test necesarios para que se pueda diagnosticar.
Suelen pasar entre 5-8 años desde el momento en el que el paciente nota los primeros sintomas de su enfermedad, hasta que llega el diagnóstico correcto.
CUADRO CLÍNICO:
-Atetosis
-Temblor
-Tics
-Sacudidas miclónicas.
FUENTE:
- http://www.imbiomed.com.mx/1/1/articulos.php?method=showDetail&id_revista=91&id_seccion=2834&id_ejemplar=5760&id_articulo=56924
-http://www.ecured.cu/index.php/S%C3%ADndrome_de_Meige
-Distonía laríngea.
-Faríngea
-Oromandibular
-Distonía bucolingual.
CAUSA:
Actualmente es de origen desconocido.
DIAGNÓSTICO:
Es frecuente que se produzcan errores de diagnóstico, pues no se poséen las pruebas o test necesarios para que se pueda diagnosticar.
Suelen pasar entre 5-8 años desde el momento en el que el paciente nota los primeros sintomas de su enfermedad, hasta que llega el diagnóstico correcto.
CUADRO CLÍNICO:
-Atetosis
-Temblor
-Tics
-Sacudidas miclónicas.
FUENTE:
- http://www.imbiomed.com.mx/1/1/articulos.php?method=showDetail&id_revista=91&id_seccion=2834&id_ejemplar=5760&id_articulo=56924
-http://www.ecured.cu/index.php/S%C3%ADndrome_de_Meige
Síndrome de Tolosa - Hunt
Es una enfermedad caracterizada por la presencia de dolor en la órbita relacionado con la oftalmoplejia ipsilateral.
Está provocada por una inflamación granulomatosa no específica del seno cavernoso.

Está provocada por una inflamación granulomatosa no específica del seno cavernoso.
CAUSAS:
La causa sigue siendo desconocida. Sin embargo, su sintomatología se explica por la inflamación granulomatosa inespecífica del seno cavernoso, la fisura orbitaria superior o la órbita.
Hipertricosis lanuginosa congénita
¡¡Buenos días de nuevo!! Hoy hablaremos del tema de la hipertricosis lanuginosa congénita. Hipertricosis se refiere al crecimiento de pelo en el cuerpo de forma excesiva. En el caso de esta alteración, el vello crecerá por todo el cuerpo excepto en las palmas de las manos y en las plantas de los pies, y puede llegar a medir hasta 25 centímetros.
 |
| Fuente: www.lasenfermedadesnuncavistas.blogspot.com |
Síndrome de DiGeorge
Es una enfermedad causada por la delección de una parte del cromosoma número 22. Sigue un patrón de herencia de tipo autosómico dominante.
Tiene una ampla variedad de síntomas, aunque no todos se dan en un mismo paciente. Normalmente se suele encontrar infecciones recurrentes, defectos cardíacos, situaciones de hipocalcemia, cosntitución facial característica y problemas de paladar.
Tiene una ampla variedad de síntomas, aunque no todos se dan en un mismo paciente. Normalmente se suele encontrar infecciones recurrentes, defectos cardíacos, situaciones de hipocalcemia, cosntitución facial característica y problemas de paladar.
Pericarditis constrictiva
Es una inflamación prolongada del pericardio, con procesos de engrosamiento, cicatrización y rigidez muscular, es decir, una especie de contractura.

Síndrome de Treacher Collins
Es una enfermedad rara que sólo se da en 1 de cada 50.000 nacimientos y cuyos rasgos aparentes resultan, por desgracia, tremendamente llamativos. Se basa fundamentalmente en defectos craneofaciales tales como la ausencia de pómulos.
Su causa es la mutación de un gen del cromosoma 5 y se puede dar bien por la transmisión hereditaria del gen defectuoso o bien de manera espontánea, provocando en cualquiera de los casos la correcta formación de los huesos del cráneo, mejillas y mandíbula.
Otras deformidades comunes con el peligroso bloqueo de las vías aéreas, la hipoplasia (desarrollo incompleto de los huesos malares) o el paladar hendido.
Su causa es la mutación de un gen del cromosoma 5 y se puede dar bien por la transmisión hereditaria del gen defectuoso o bien de manera espontánea, provocando en cualquiera de los casos la correcta formación de los huesos del cráneo, mejillas y mandíbula.
Otras deformidades comunes con el peligroso bloqueo de las vías aéreas, la hipoplasia (desarrollo incompleto de los huesos malares) o el paladar hendido.
Malformación de Arnold Chiari
Es una anomalía cerebral que afecta al cerebelo. De esta forma, el cerebelo sobresalirá y ocupará parte del espacio que normalmente ocupa la médula espinal.

CAUSAS:
En la mayoría de los casos se trat de una anomalía congénita, es decir, que el niño la desarrollo en el útero materno y ya la poseía en el momento del nacimiento.
En otros casos, puede ser adquirida, es decir, que se va desarrollando durante el transcurso de la vida del paciente, debido a factores relacionados con trastornos que alteran el desarrollo del cerebro, médula espinal y los huesos.
Se cree que puede ser de tipo hereditario.
Síndrome de Bloom
Se trata de una patología extremadamente rara que se caracteriza por la hipersensibilidad a la luz solar, lo que lleva a la aparición de un "rash" eritomatoso en la zona de las mejillas o el dorso de las manos. Otros indicadores son las telangiectasias, voz aguda, baja estatura, cara larga y estrecha, nariz y orejas prominentes, etc.
Sigue un patrón de herencia autosómica-recesiva.y está asociada a mutaciones del gen BLM, que codifica a una proteína de la familia de las helicasas (implicadas en la replicación y transcripción del ADN).
Sigue un patrón de herencia autosómica-recesiva.y está asociada a mutaciones del gen BLM, que codifica a una proteína de la familia de las helicasas (implicadas en la replicación y transcripción del ADN).
Hemiatrofia facial. Síndrome de Parry - Romberg
Es un síndrome de tipo neurocutáneo de etilogía desconocida. Entre sus principales características destacan:
Atrofia de tejido blandos y algunas veces de hueso. En ocasiones, esta atrofia se extiende a las extremidades y puede dar lugar a complicaciones oftamológicas y neurológicas.

Atrofia de tejido blandos y algunas veces de hueso. En ocasiones, esta atrofia se extiende a las extremidades y puede dar lugar a complicaciones oftamológicas y neurológicas.
CARACTERÍSTICAS CLÍNICAS:
-Hemiatrofia facial de grasa, piel, tejido conectivo, músculo y/o hueso.
-Hemiatrofia de brazo, tronco o pierna.
-Atrofia de la lengua.
-Anormalidades en dientes y dificultades para abrir la boca.
-Migraña, dolor facial.
-Epilepsia.
-Anormalidades oculares.
-Epilepsia.
-Vitíliga
-Anormalidades del cerebro visibles en resonancia magnética.
Still del adulto
Buenos días! Hoy trataremos la enfermedad del STILL DEL ADULTO.

Lo que ocurre cuando se padece esta enfermedad es que aparecen fiebres altas, sarpullido y dolor en las articulaciones que puede terminar en artritis crónica. Se trata de la versión severa de la artritis idiopática juvenil.
La causa es totalmente desconocida, hay hipótesis sobre virus o agentes infecciosos pero no se ha llegado a una conclusión evidente.
lunes, 15 de diciembre de 2014
Anemia de Diamond-Blackfan o ADB
¡¡Buenos días a todos!! Hoy, y empezando la semana, vamos a dedicar una de nuestras entradas diarias a la anemia de Diamond-Blackfan.

Como ya sabréis, en la anemia se encuentra afectados la hemoglobina y los glóbulos rojos. Pues en la anemia de Diamond-Blackfan (ADB de aquí en adelante) también habrá un número bajo de glóbulos rojos, ya que la médula ósea no producirá los suficientes.
La incidencia de esta enfermedad, en términos anuales, está alrededor de un caso por cada 150000 nacimientos. Por lo tanto, su incidencia se encuentra dentro de los parámetros necesarios para incluir una enfermedad dentro del grupo de las enfermedades raras.
La incidencia de esta enfermedad, en términos anuales, está alrededor de un caso por cada 150000 nacimientos. Por lo tanto, su incidencia se encuentra dentro de los parámetros necesarios para incluir una enfermedad dentro del grupo de las enfermedades raras.
Enfermedad de Raynaud
Buenos días a todos!!
Hoy hablaremos sobre la ENFERMEDAD DE RAYNAUD.
Se trata de una enfermedad que afecta a los vasos sanguíneos situados en los dedos de manos y pies. Cuando la persona siente frío o estrés los vasos se estrechan y no dejan llegar la sangre a esta zona. Este fenómeno hace que los dedos se vuelvan blanquecinos o azulados. En casos extremos puede haber muerte celular, pero si la sangre vuelve a fluir la piel se enrojece y aparece pálpito y hormigueo.

Existen dos tipos diferentes según el origen de la enfermedad. El fenómeno de Raynaud primario es aquel que no tiene una causa conocida, en cambio el fenómeno de Raynaud secundario ocurre asociado a otras enfermedades.
Algunas de ellas son la ateroesclerosis, artritis y enfermedades autoinmunes, pero también el tabaquismo o diversos fármacos propician la aparición de la enfermedad de Raynaud.
domingo, 14 de diciembre de 2014
Síndrome de McCune-Albright
¡Buenos días de domingo! Hoy en esta entrada hablaremos del síndrome de McCune-Albright. Esta enfermedad afectará principalmente a los huesos y a la piel, y afectará también al inicio de la pubertad.
Si recordáis, ayer en la entrada en la que hablábamos del síndrome de Apert, decíamos que podía ser heredada o producirse por una mutación espontánea, que era lo más frecuente. Sin embargo, en la enfermedad de la que hablaremos hoy, no hay posibilidad de que se transmita a la descendencia, produciéndose única y exclusivamente por mutaciones "de novo".
La mutación se produce en el gen GNAS1 del cromosoma 20, y este gen es el encargado de producir una proteína cuya función se centra en las disposiciones musculo-esqueléticas y también en las hormonas, lo que explica que al estar alterado afecte a los huesos, a la piel, y a la pubertad.
La mutación que se produce en este caso se denomina mosaicismo. Esto es, que puede producirse en varias células o en todas, y su distribución será al azar.
La mutación se produce en el gen GNAS1 del cromosoma 20, y este gen es el encargado de producir una proteína cuya función se centra en las disposiciones musculo-esqueléticas y también en las hormonas, lo que explica que al estar alterado afecte a los huesos, a la piel, y a la pubertad.
La mutación que se produce en este caso se denomina mosaicismo. Esto es, que puede producirse en varias células o en todas, y su distribución será al azar.
Síndrome de Barret
Se define como un trastorno a través del cual el revestimiento del esófago presenta daño a causa del ácido gástrico y se vuelve similar al del estómago.

CAUSAS:
Cuando comemos el alimento pasa de la garganta al estómago a través del esofágo. Un anillo de fibras musculares situado en la parte inferior de este, se encargará de impidir que los contenidos estomacales se devuelvan.
Si estas fibras no se cierra herméticamente, se puede filtrar un ácido áspero hacia el esófago, pudiendo causar daño tisular con el tiempo.
Ocurre más frecuentemente en hombres que en mujeres.
Enfermedad de Weil
Es una infección poco habitual y de carácter grave, que tiene lugar cuando se contacta con la bacteria Leptospira.
CAUSAS:
Esta bacteria se encuentra en aguas dulces que han podido ser contaminadas por medio de la orina de los animales. Tal infección ocurre en climas cálidos.
Normalmente no se propaga de una persona a otra.
FACTORES DE RIESGO:
- Exposición ocupacional: Granjeros, cazadores,...
- Actividades recreativas como nadar en aguas dulces.
- Exposición en el hogar: Mascotas, ganado doméstico,...
CAUSAS:
Esta bacteria se encuentra en aguas dulces que han podido ser contaminadas por medio de la orina de los animales. Tal infección ocurre en climas cálidos.
Normalmente no se propaga de una persona a otra.
FACTORES DE RIESGO:
- Exposición ocupacional: Granjeros, cazadores,...
- Actividades recreativas como nadar en aguas dulces.
- Exposición en el hogar: Mascotas, ganado doméstico,...
Neurofibromatosis
Hoy hablaremos de la NEUROFIBROMATOSIS.
Se trata de una anomalía genética del sistema nervioso en la cual las células crecen incontrolablemente formando tumoraciones en los nervios. Normalmente éstos son benignos pero también existe la posibilidad de que se conviertan en cáncer.
La causa es genética, bien por herencia de padres a hijos, o bien por mutación de los genes.
Tipos
Existen tres tipos de Neurofibromatosis. El tipo 1 afecta a la piel y a los huesos provocando deformidades. Suele comenzar sus síntomas en el nacimiento. El tipo 2 se caracteriza por pérdida de oído, zumbidos o problemas de equilibrio. Comienza en la adolescencia. Y la schwanomatosis es el tipo más anómalo y se caracteriza por el dolor intenso.
Síntomas
Neurofibromatosis 1: dolor en el área dañada así como deterioro nervioso, manchas café con leche, ceguera, convulsiones, pecas, neurofibromas plexiformes bajo la piel y neurofibromas nodulares en la piel.

sábado, 13 de diciembre de 2014
Síndrome de Apert
¡Buenas noches! Hoy vamos a hablar del síndrome de Apert.
El síndrome de Apert va a afectar generalmente a la cara y la cabeza, aunque también puede afectar a manos y pies. Se debe a que los huesos del cráneo se van a unir y cerrar de forma prematura; esto se conoce como craneosinostosis. Otra de las enfermedades que se puede producir también por craneosinostosis es la enfermedad de Crouzon, de la cual se publicó información en este blog hace unos días.
El síndrome de Apert va a afectar generalmente a la cara y la cabeza, aunque también puede afectar a manos y pies. Se debe a que los huesos del cráneo se van a unir y cerrar de forma prematura; esto se conoce como craneosinostosis. Otra de las enfermedades que se puede producir también por craneosinostosis es la enfermedad de Crouzon, de la cual se publicó información en este blog hace unos días.
 |
| Fuente: www.escuela.med.puc.cl |
Enfermedad de Bowen
Se trata de una enfermedad basada en una lesión de tipo precancerosa,que afecta a las capas más superficiales de la piel.
La mayoría de los casos aparece debido a la exposición crónica al sol, por lo que suele aparecer en forma de lesión en la cara, en el dorso de las manos y en las piernas:

La mayoría de los casos aparece debido a la exposición crónica al sol, por lo que suele aparecer en forma de lesión en la cara, en el dorso de las manos y en las piernas:
Es frecuente que se produzca en personas adultas y que además tengan la piel clara.
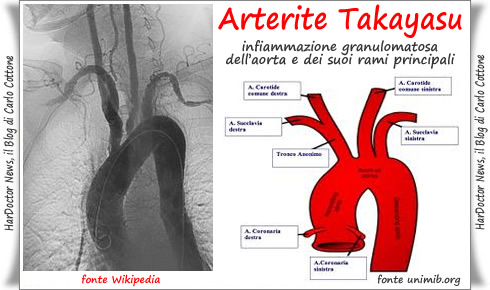
Arteritis de Takayasu
Consiste en una inflamación de la arteria aorta y sus ramificaciones:
- Fiebre.
- Mareo.
- Dolor muscular o articular.
- Erupción cutánea.
- Sudores fríos.
- Cambios en la visión.
- Pérdida depeso.
CAUSAS:
Su causa se desconoce. Esta patología tiene lugar princepalmente en niños y en mujeres de menos de 40 años de edad, siendo más común en personas asiáticas y africanas.
Parece ser que se trata de una enfermedad de tipo autoinmunitario, por lo que el sistema inmunitario atacará a los tejidos sanos por error.
SÍNTOMAS:
- Debilidad o dolor en los brazos.
- Dolor torácico.
- Fatiga.- Fiebre.
- Mareo.
- Dolor muscular o articular.
- Erupción cutánea.
- Sudores fríos.
- Cambios en la visión.
- Pérdida depeso.
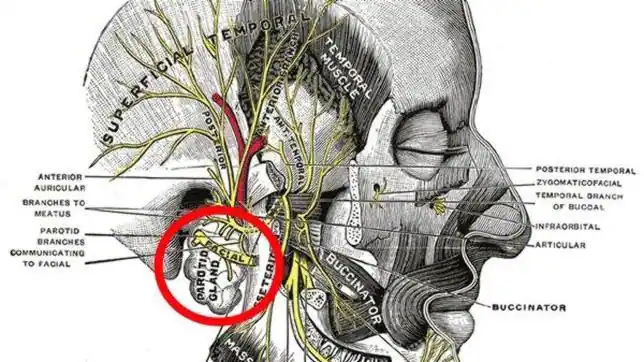
Sudoración gustativa
Es frecuente que todos y cada uno de nosotros, comencemos a sudar tras la ingestión de alimentos calientes o picantes, pues estos son capaces de elevar la temperatura corporal.
En cambio, un número más reducido de personas, sudan al tomar o pensar en cualquier tipo de alimento. Nos encontramos entonces ante un caso de sudoración gustativa o síndrome de Frey.
En muchos casos, este problema surge después de un traumatismo o cirugía de las glándulas parótidas, encargadas de la salivación.
En cambio, un número más reducido de personas, sudan al tomar o pensar en cualquier tipo de alimento. Nos encontramos entonces ante un caso de sudoración gustativa o síndrome de Frey.
En muchos casos, este problema surge después de un traumatismo o cirugía de las glándulas parótidas, encargadas de la salivación.
La salivación es un proceso relacionado con el proceso de ingestión y que aparece en la prepación para la difestión y en la masticación.
Enfermedad de Marek
También es conocida con el nombre de parálisis de las gallinas. Es una enfermedad viral y neoplástica. Está causada por un virus herpes alfa.
Es una enfermedad caracterizada por la presencia de linfomas de células T, así como por la infiltración de nervios y órganos por medio de los linfocitos.
Existen varios síndromes identificados que se manifiestan durante un proceso de infección de la enfermedad de Marek:
- Enfermedad de Marek clásica.
- Enfermedad de Marek aguda.
- Linfomatesis ocular.
- Enfermedad de Marek cutánea.
- Arteriosclerosis.
- Inmunodepresión.
Diagnóstico:
Hay que verificar la inflamación de los nervios, especialmente del nervio ciático. También conviene presta atención a la aparición de nódulos en los órganos internos.
Fuente: http://es.wikipedia.org/wiki/Enfermedad_de_Marek
Es una enfermedad caracterizada por la presencia de linfomas de células T, así como por la infiltración de nervios y órganos por medio de los linfocitos.
Existen varios síndromes identificados que se manifiestan durante un proceso de infección de la enfermedad de Marek:
- Enfermedad de Marek clásica.
- Enfermedad de Marek aguda.
- Linfomatesis ocular.
- Enfermedad de Marek cutánea.
- Arteriosclerosis.
- Inmunodepresión.
Diagnóstico:
Hay que verificar la inflamación de los nervios, especialmente del nervio ciático. También conviene presta atención a la aparición de nódulos en los órganos internos.
Fuente: http://es.wikipedia.org/wiki/Enfermedad_de_Marek
Enfermedad de Pick
Es una forma atípica de demencia. Es permanente.
Se diferencia del Alzheimer en que la enfermedad de Pick afecta a unas áreas determinadas del cerebro.
CAUSAS:
Las personas que padecen esta patologís poséen unos cuerpos y células de Pick, dentro de las neuronas que se encuentran en las zonas afectadas del cerebro. Tales cuerpos tienen una forma anormal de la proteína TAU, proteína que se encuentra presente en todas las neuronas.
La causa de por qué esta proteína se ve alterada, se desconoce. Sin embargo, se sabe que distintos genes que pueden verse alterados pueden provocar esta patología y que a veces se hereda de padres a hijos. Es una enfermedad muy poco común y suele empezar a desarrollarse entre los 40-60 años.
Se diferencia del Alzheimer en que la enfermedad de Pick afecta a unas áreas determinadas del cerebro.
CAUSAS:
Las personas que padecen esta patologís poséen unos cuerpos y células de Pick, dentro de las neuronas que se encuentran en las zonas afectadas del cerebro. Tales cuerpos tienen una forma anormal de la proteína TAU, proteína que se encuentra presente en todas las neuronas.
La causa de por qué esta proteína se ve alterada, se desconoce. Sin embargo, se sabe que distintos genes que pueden verse alterados pueden provocar esta patología y que a veces se hereda de padres a hijos. Es una enfermedad muy poco común y suele empezar a desarrollarse entre los 40-60 años.
Hipertensión pulmonar
Buenos días a todos!! Hoy he escogido como tema la HIPERTENSIÓN PULMONAR.
Lo que ocurre cuando se padece esta enfermedad es que las arterias que se encuentran en los pulmones se hacen más estrechas de lo normal y por lo tanto el flujo sanguíneo que pueden transportar es menor. La presión que ejerce la sangre en esta zona se incrementa, de modo que el corazón necesita más fuerza para bombearla.
Con el paso del tiempo el lado derecho del corazón (la zona que se encarga de bombear sangre a los pulmones) aumenta de tamaño, esto se conoce como insuficiencia cardíaca del lado derecho o cor pulmonae.

Etiología
Las causas de la hipertensión pulmonar son muy diversas. Puede ocurrir por enfermedades que afectan a los pulmones, por anomalías congénitas del corazón, embolias pulmonares, insuficiencia cardíaca o fármacos.
Cuando la causa es desconocida se denomina hipertensión pulmonar idiopática.
viernes, 12 de diciembre de 2014
Progeria o síndrome de Hutchinson-Gilford
¡¡Buenas tardes!! Hoy empezando el fin de semana también vamos a publicar entradas. Hablaremos aquí del síndrome de Hutchinson-Gilford o también llamado progeria. Quizás recordéis una entrada de este blog en la que hablábamos del síndrome de Werner; si no es así, os recuerdo yo un poco de que iba. Se trataba de la aparición de rasgos de envejecimiento en la tercera década de la vida. Esto va a tener algo en común, y es que en esta enfermedad también aparecen rasgos de envejecimiento, pero en este caso, sucede durante la infancia, y de hecho, la esperanza de vida que tienen estas personas a menudo no va a superar los 30 años.
La causa de esta enfermedad es desconocida, pero sin embargo una hipótesis que se postula es que se debe a una mutación en un gen, cuya función es la supresión del fenotipo de envejecimiento. Al producirse la mutación, no cumple su función y se produce la enfermedad. La mayoría de los casos de esta enfermedad se produce de forma esporádica, lo que conocemos como las mutaciones "de novo". Además, la incidencia es un dato importante en este caso, ya que aproximadamente afecta a una de cada ocho millones de personas.
La causa de esta enfermedad es desconocida, pero sin embargo una hipótesis que se postula es que se debe a una mutación en un gen, cuya función es la supresión del fenotipo de envejecimiento. Al producirse la mutación, no cumple su función y se produce la enfermedad. La mayoría de los casos de esta enfermedad se produce de forma esporádica, lo que conocemos como las mutaciones "de novo". Además, la incidencia es un dato importante en este caso, ya que aproximadamente afecta a una de cada ocho millones de personas.
Hepatitis crónica autoinmune
Buenas tardes!! Hoy hablaremos sobre la HEPATITIS CRÓNICA AUTOINMUNE.
Se trata de una enfermedad crónica en la que el hígado se inflama por el ataque del sistema inmunitario.

Etiología
Su etiología es desconocida pero las hipótesis apuntan a anomalías genéticas en los antígenos de histocompatibilidad HLA-B8 y HLA-DR3. Lo que si se sabe es que afecta en mayor medida al sexo femenino, sobre todo a niñas y mujeres jóvenes.
Síntomas
Los síntomas que se manifiestan pueden confundirse con una hepatitis aguda. Aparece fiebre, náuseas, vómitos, ictericia pero también se muestran otros como el abdomen distendido, color oscuro en la orina, heces claras o fatiga e inapetencia.
jueves, 11 de diciembre de 2014
Enfermedad de Kawasaki
¡¡Buenos días a todos una vez más!!
Hoy hablaremos de la enfermedad de Kawasaki. Estamos ante una enfermedad rara que en un país como EEUU por ejemplo, se estima que solo se da en 19 de cada 100000. En Europa no es muy frecuente, y afecta alrededor de 5 de cada 100000 personas. También se da con más frecuencia en otro país como es Japón, donde afecta aproximadamente a 100 de cada 100000 personas. Además, estas personas serán niños, ya que esta enfermedad les afecta a ellos. Y a mayores, podemos decir que frecuentemente se trata de niños de menos de cinco años.
Seguramente estáis pensando: "¡Basta ya de datos! ¿En qué consiste esta enfermedad?" Pues ahora vamos a explicarlo. Esta enfermedad va a afectar principalmente a mucosas, a la piel y a los ganglios linfáticos, implicando la inflamación de los vasos sanguíneos. Quizás con esto no os aclare mucho, pero ahora veremos los síntomas que pueden darse en esta enfermedad y podremos ubicar un poco en qué consiste. Además, la causa de la enfermedad de Kawasaki es desconocida.
Hoy hablaremos de la enfermedad de Kawasaki. Estamos ante una enfermedad rara que en un país como EEUU por ejemplo, se estima que solo se da en 19 de cada 100000. En Europa no es muy frecuente, y afecta alrededor de 5 de cada 100000 personas. También se da con más frecuencia en otro país como es Japón, donde afecta aproximadamente a 100 de cada 100000 personas. Además, estas personas serán niños, ya que esta enfermedad les afecta a ellos. Y a mayores, podemos decir que frecuentemente se trata de niños de menos de cinco años.
Seguramente estáis pensando: "¡Basta ya de datos! ¿En qué consiste esta enfermedad?" Pues ahora vamos a explicarlo. Esta enfermedad va a afectar principalmente a mucosas, a la piel y a los ganglios linfáticos, implicando la inflamación de los vasos sanguíneos. Quizás con esto no os aclare mucho, pero ahora veremos los síntomas que pueden darse en esta enfermedad y podremos ubicar un poco en qué consiste. Además, la causa de la enfermedad de Kawasaki es desconocida.
Esclerodermia
Hoy hablaremos de la ESCLERODERMIA. Se trata de una enfermedad autoinmune en la que el organismo arremete contra el tejido sano.
Tipos
Existen dos tipos:
La esclerodermia localizada, que se manifiesta en manos y cara. Es muy raro que se se extienda a otras zonas.
Y la esclerodermia sistémica que aparece en áreas extensas de la piel, incluso en órganos principales (corazón, pulmones o riñones).
Causa
La causa de esta enfermedad es desconocida. Se trata de un modelo poliegénico, ya que son varios genes los que participan en esta anomalía y también influyen las condiciones ambientales.
Síntomas
Cutáneos: dedos y manos azulados o fríos (fenómeno Raynaud), alopecia, color facial anormal, rigidez en las extremidades superiores, quistes blancos subcutáneos que a veces supuran un líquido blanco, ulceraciones en las puntas de los dedos y piel de la cara tensa.

miércoles, 10 de diciembre de 2014
Síndrome de Felty
¡¡Buenas tardes!! Hoy hablaremos del síndrome de Felty. Este es una enfermedad que cursa con tres patologías principalmente. Estas son: artritis reumatoide, aumento del tamaño del bazo (esplenomegalia) y neutropenia (recuento bajo de glóbulos blancos).
 |
| Fuente: www.effectivehealthcare.ahrq.gov |
 |
| Fuente: www.keckmedicine.adam.com |
Epidermólisis bullosa
Buenas! Otro día más os hablaremos de algún tipo de enfermedad clasificada como rara. Yo os hablaré hoy de la EPIDERMÓLISIS BULLOSA o AMPOLLOSA (EB).

Se trata de un conjunto de afecciones dérmicas hereditarias caracterizadas por la aparición de ampollas u otras lesiones cutáneas tras una simple rozadura o contusión.
Existen 30 tipos de epidermólisis ampollosa pero pueden agruparse principalmente en tres tipos.
La EB Simplex está caracterizada por la cisura de la epidermis con una cicatrización sin pérdida de continuidad.
martes, 9 de diciembre de 2014
Botulismo
¡Buenas tardes de nuevo! Hoy hablaremos del botulismo. El botulismo es una enfermedad que se origina por una bacteria: clostidrium botulinum. Esta bacteria la podemos captar de dos formas: mediante esporas, bien a través de la ingestión de alimentos, o bien a través de la penetración en heridas. A mayores existe también el botulismo del lactante que suele producirse al ingerir miel. Con el paso del tiempo se va a producir la toxina, de la cual aunque se ingiera una mínima cantidad, la intoxicación puede ser grave. En esta imagen os dejo un poco resumido lo que acabo de explicar, y además aparecen las vías de alimentos mediante las cuales es más frecuente llegar a tener botulismo.
 |
| Fuente: www.edgarmelendez.wordpress.com Podéis ampliar la imagen haciendo clic en ella. |
Cistitis intersticial
Buenos días! Hoy vamos a hablar de la CISTITIS INTERSTICIAL.

Se trata de una enfermedad crónica que se caracteriza por la sensación de dolor, presión o ardor en la zona pélvica, concretamente en la vejiga, por ello también es conocida como el síndrome de la vejiga dolorosa.
lunes, 8 de diciembre de 2014
Enfermedad de Buerger
Hoy hablaremos de la Enfermedad de BUERGER, también conocida como Tromboangeítis Obliterante.

Se trata de una enfermedad que provoca la obstrucción de los vasos sanguíneos de pies y manos debido a una inflamación e hinchazón de los mismos.
El influjo sanguíneo puede terminar en la formación de coágulos e incluso en gangrena. Cuando esto ocurre podría ser necesaria la amputación.
Enfermedad de Batten
¡¡Hola a todos los que nos leéis!!
La enfermedad de Batten es una enfermedad mortal del sistema nervioso que se produce por el mal funcionamiento de algunos genes y por lo tanto, la mala producción y uso de proteínas. Esto hace que se acumulen proteínas y grasas en forma de lipopigmentos principalmente en el cerebro, aunque también puede presentarse en otras partes, como los ojos.
Esta enfermedad se conoció por primera vez en 1903. Desde entonces, no se han dado muchos casos y su incidencia en la actualidad sigue sin ser alta, de ahí que se considere una enfermedad rara. Al igual que ayer llevemos el ejemplo a un país como EEUU, donde de cada 100000 personas, solo entre dos o cuatro llegan a tener esta enfermedad.
Esta enfermedad se conoce también como la forma juvenil de NLC (ceroidolipofuscinosis neuronales), aunque existen más tipos como la infantil, la infantil tardía y la adulta.
domingo, 7 de diciembre de 2014
Citomegalovirus
Hoy hablaremos del CITOMEGALOVIRUS.
Se trata de una forma de herpesvirus, concretamente el Human herpesvirus 5 o HHV-5.
Este virus es común y por lo tanto cualquiera puede estar afectado por él sin saberlo porque apenas aparecen síntomas si la persona está sana, simplemente estaría el virus latente. Pero el problema existe si la persona que lo contrae está inmunodeprimida debido a una enfermedad o por tratamientos, o si dicha persona está embarazada.

Contagio
La forma de propagación es a través de fluidos corporales: sangre, saliva, orina, semen, fluidos vaginales, heces, leche materna, etc. También puede ocurrir el contagio cuando tienen lugar transfusiones o trasplantes.
Los bebés pueden infectarse durante el embarazo o después del parto. También es fácil que se contagien posteriormente en la guardería o centros escolares.
Síndrome de Noonan
¡¡Buenos días de domingo!! La entrada de hoy se va a centrar en el síndrome de Noonan.
El síndrome de Noonan es una enfermedad que impide que varias partes del cuerpo se desarrollen de manera normal. Esto ha de saber diferenciarse del síndrome de Turner, ya que pueden llegar a confundirse. De hecho, a esta enfermedad a veces también se le denomina con otros nombres como "síndrome familiar de Turner".
Es una enfermedad hereditaria que se produce por una mutación en el cromosoma 12, y principalmente los genes afectados son el KRAS, PTPN11, RAF1 y SOS1. A pesar de esto y aunque puede transmitirse hereditariamente, también pueden darse casos de novo. Cuando se transmite a la descendencia sigue un patrón autosómico dominante, y en general, suele ser por vía materna.
Se agrupa junto con las enfermedades raras porque la incidencia es baja, de hecho, suele ser de un caso por cada 2500 nacimientos.
Los síntomas que se pueden ver con esta enfermedad afecta a varias partes del cuerpo, como ya introducíamos al principio.
El síndrome de Noonan es una enfermedad que impide que varias partes del cuerpo se desarrollen de manera normal. Esto ha de saber diferenciarse del síndrome de Turner, ya que pueden llegar a confundirse. De hecho, a esta enfermedad a veces también se le denomina con otros nombres como "síndrome familiar de Turner".
Es una enfermedad hereditaria que se produce por una mutación en el cromosoma 12, y principalmente los genes afectados son el KRAS, PTPN11, RAF1 y SOS1. A pesar de esto y aunque puede transmitirse hereditariamente, también pueden darse casos de novo. Cuando se transmite a la descendencia sigue un patrón autosómico dominante, y en general, suele ser por vía materna.
Se agrupa junto con las enfermedades raras porque la incidencia es baja, de hecho, suele ser de un caso por cada 2500 nacimientos.
Los síntomas que se pueden ver con esta enfermedad afecta a varias partes del cuerpo, como ya introducíamos al principio.
sábado, 6 de diciembre de 2014
Psitacosis
¡¡Buenos días de nuevo!! Hoy estoy aquí para hablaros de la psitacosis. Todos estos días hemos hablado de enfermedades raras que afectaban a humanos y la mayoría se transmitían de forma hereditaria. Esta enfermedad difiere un poco con respecto a las anteriores de las que ya hemos hablado, ya que sí que afecta a humanos pero sin embargo, la fuente de infección es diferente ya que quien lo puede transmitir son aves que pertenezcan a las familias de loros principalmente, y también pueden ser pavos, palomas u otras especies.
 |
| Fuente: www.diariouno.com.ar |
Quiste de Tarlov
Buenos días a todos!! Hoy vamos a hablar de los QUISTES DE TARLOV.
Se trata de unas masas que se forman en la duramadre y aracnoides, las dos capas más profundas de ls meninges, en concreto como dilatación de la duramadre. Se componen de líquido cefalorraquídeo y poseen un pedículo para comunicarse con el espacio subaracnoideo espinal. Están situados en la región sacra y lumbar y afectan a las raíces nerviosas debido a la compresión de los quistes.

viernes, 5 de diciembre de 2014
Síndrome de Miller-Dieker
¡¡Buenas noches a todos!!
Hoy esta entrada la vamos a ocupar hablando del síndrome de Miller-Dieker. De nuevo, se trata de una enfermedad rara aunque en esta no podemos concretar la incidencia en la población, ya que es desconocida, pero el número de casos es bajo.
Además, es una enfermedad hereditaria que se produce por una mutación en el cromosoma 17. Pero como ya apuntamos en otras ocasiones, esta enfermedad puede producirse por una mutación espontánea o de novo o por la herencia, en cuyo caso sigue un patrón autosómico dominante.
Hoy esta entrada la vamos a ocupar hablando del síndrome de Miller-Dieker. De nuevo, se trata de una enfermedad rara aunque en esta no podemos concretar la incidencia en la población, ya que es desconocida, pero el número de casos es bajo.
Además, es una enfermedad hereditaria que se produce por una mutación en el cromosoma 17. Pero como ya apuntamos en otras ocasiones, esta enfermedad puede producirse por una mutación espontánea o de novo o por la herencia, en cuyo caso sigue un patrón autosómico dominante.
Epispadias
Buenos días! Hoy hablaremos sobre la enfermedad rara de EPISPADIAS.
Se trata de una anomalía congénita situada en el orificio uretral en la que el conducto uretral no está completo y la orina sale del cuerpo por un lugar incorrecto.
La causa es desconocida pero se relaciona con un error en el desarrollo de la formación de los genitales externos. Este problema también puede incluir defectos en la vejiga, uretra e intestino grueso. Cuando la epispadias está acompañado con que la vejiga emerge a través del abdomen se denomina extrofia vesical.

Extrofia vesical
jueves, 4 de diciembre de 2014
Síndrome de fatiga crónica
¡¡Buenas noches a todos!!
Hoy vamos a hablar un poco acerca del síndrome de la fatiga crónica. Esta es una enfermedad que a veces puede confundirse con la fibromialgia por la similitud de síntomas, pero sin embargo, no son lo mismo.
A día de hoy todavía no se sabe con certeza la causa, pero aún así se desconfía de dos factores. Por una parte podría ser que el sistema nervioso se encontrase inflamado, y por otra parte, podría deberse a un virus, el virus VEB. Además, pueden influir en esto factores como la edad, el estrés al que esté sometido la persona o la actuación de diferentes factores ambientales sobre la persona.

Aproximadamente, afecta a un 0,5% de la población global, y afecta más a mujeres que a hombres.
Hoy vamos a hablar un poco acerca del síndrome de la fatiga crónica. Esta es una enfermedad que a veces puede confundirse con la fibromialgia por la similitud de síntomas, pero sin embargo, no son lo mismo.
A día de hoy todavía no se sabe con certeza la causa, pero aún así se desconfía de dos factores. Por una parte podría ser que el sistema nervioso se encontrase inflamado, y por otra parte, podría deberse a un virus, el virus VEB. Además, pueden influir en esto factores como la edad, el estrés al que esté sometido la persona o la actuación de diferentes factores ambientales sobre la persona.
Aproximadamente, afecta a un 0,5% de la población global, y afecta más a mujeres que a hombres.
Pénfigo foliáceo
Hoy hablaremos sobre el PÉNFIGO FOLIÁCEO.

Cuando nos referimos al pénfigo estamos haciendo alusión a un conjunto de enfermedades auto inmunes que afectan a la piel.
Lo que ocurre es que el sistema inmune produce anticuerpos que atacan a las células sanas de piel y mucosas. De esta forma las células dérmicas se separan, entre las capas de la piel hay acumulación de líquido y aparecen ampollas.
Linfoma de Burkitt
El linfoma de Burkitt es un tipo de linfoma no Hodgkin de crecimiento muy rápido. Incialmente se descubrió en niños en alguans zonas de África, aunque también está presente en Estados Unidos.
El tipo africano está vinculado al virus de Epstein - Barr, mientras que el estadounidense no lo está.
Las personas que padecen VIH tienen un mayor riesgo de sufrir esta patología. Tambíen cabe mencionar, que se observa con mayo frecuencia en hombres.
En cuanto a las manifestaciones clínicas, se presenta como una inflamación de los ganglios linfáticos del cuello, la ingle o puede que includo en la zona de debajo del brazo. Frecuentemente esta inflamación es de tipo indolora, sin embargo, pueden crecer rápidamente.
En los tipos más comunes observados en los Estados Unidos, el cáncer suele comenzar por la zona abdominal, aunque también puede empezar por otras zonas como los ovarios, testículos, cerebro o líquido cefalorraquídeo.
El tipo africano está vinculado al virus de Epstein - Barr, mientras que el estadounidense no lo está.
Las personas que padecen VIH tienen un mayor riesgo de sufrir esta patología. Tambíen cabe mencionar, que se observa con mayo frecuencia en hombres.
En cuanto a las manifestaciones clínicas, se presenta como una inflamación de los ganglios linfáticos del cuello, la ingle o puede que includo en la zona de debajo del brazo. Frecuentemente esta inflamación es de tipo indolora, sin embargo, pueden crecer rápidamente.
En los tipos más comunes observados en los Estados Unidos, el cáncer suele comenzar por la zona abdominal, aunque también puede empezar por otras zonas como los ovarios, testículos, cerebro o líquido cefalorraquídeo.
Enfermedad de Lafora
La enfermedad de Lafora, es una forma de epilepsia mioclónica progresiva y se presenta principalmente entre los países del sur de Europa.
La incidencia de esta patología es muy escasa en la población.
Se trata de una enfermedad hereditaria que se transmite de forma recesiva. Por tanto, para que esto suceda, deben aparecer dos copias del gen en la descendencia.

Los primeros síntomas y signos clínicos aparecen durante la pubertad y en la adolescencia. Generalmente se ponen de manifiesto en forma de crisis epilépticas convulsivas o puede que también en forma de crisis visuales, caracterizadas porque se describen como la visualización de luces o estrellas.
La incidencia de esta patología es muy escasa en la población.
Se trata de una enfermedad hereditaria que se transmite de forma recesiva. Por tanto, para que esto suceda, deben aparecer dos copias del gen en la descendencia.
Los primeros síntomas y signos clínicos aparecen durante la pubertad y en la adolescencia. Generalmente se ponen de manifiesto en forma de crisis epilépticas convulsivas o puede que también en forma de crisis visuales, caracterizadas porque se describen como la visualización de luces o estrellas.
miércoles, 3 de diciembre de 2014
Hemofilia
Buenas!! Esta tarde hablaremos de la HEMOFILIA.
Se trata de un trastorno hemorrágico en el que la sangre no coagula como es debido.
Causa
Cuando se produce un sangrado el organismo comienza un proceso denominado cascada de coagulación en el que intervienen distintas proteínas denominadas factores de coagulación. Cuando falta algún factor tiene lugar la hemofilia.
Es una enfermedad hereditaria en la que los hombres son los más afectados.

Tipos
HEMOFILIA A
En este tipo de hemofilia falta el factor de coagulación VIII.
La causa es un patrón hereditario recesivo ligado al cromosoma X por lo tanto el gen anormal está en el cromosoma X. Las mujeres al poseer dos cromosomas X si el gen de un cromosoma está dañado el otro cromosoma normal se hará cargo de la coagulación. En este caso la mujer se hará portadora.
Este es el motivo de que los hombres tengan mayor probabilidad de tener hemofilia de tipo a porque solo tienen un cromosoma X.
Enfermedad de Caroli
La enfermedad de Caroli, también conocida como ectasia comunicante de las vías biliares intrahepáticas, fue descrita por primera vez en el año 1958. Es una patología de tipo congénita que se caracteriza por la presencia de múltiples dilataciones saculares o quísticas de las vías biliares intrahepáticas.

En cuanto a las manifestaciones clínicas, la más habitual es el dolor de tipo abdominal, en forma de crisis de presentación recurrente. Esto se debe comúnmente a la litiaisis intrahepática, aunque también puede deberse a una litiasis vesicular asociada o a una pancreatitis aguda.
Enfermedad de Moyamoya
Es una enfermedad rara de tipo cerebrovascular que predispone a los pacientes afectados a sufrir un ictud en relación a una progresiva estenosis dela arteria carótida interna y sus principales ramas.
Debido a la reducción del flujo sanguíneo en los vasos principales de la circulación anterior del cerebro conduce al desarrollo compensatorio de circulación colateral a través de pequeñas ramas penetrantes de la arteria carótida interna, la cerebral media y la cerebral anterior.
Se forman así, prominentes canales anatomóticos, telangiectasias basales que aparecen en las angiografias como volutas de humo.
La presentación clínica variará en función de si los pacientes son niños o adultos. La principal diferencia entre ambos se centra en que la mayoría de los niños se presentan con ataques isquémicos transitorios o infartos cerebrales, mientras que en más de la mitad de los adultos la enfermedad derivarará en forma de hemorragias intracraneales, típicamente en tálamo, ganglios basales o sustancia blanca profunda.
Debido a la reducción del flujo sanguíneo en los vasos principales de la circulación anterior del cerebro conduce al desarrollo compensatorio de circulación colateral a través de pequeñas ramas penetrantes de la arteria carótida interna, la cerebral media y la cerebral anterior.
Se forman así, prominentes canales anatomóticos, telangiectasias basales que aparecen en las angiografias como volutas de humo.
La presentación clínica variará en función de si los pacientes son niños o adultos. La principal diferencia entre ambos se centra en que la mayoría de los niños se presentan con ataques isquémicos transitorios o infartos cerebrales, mientras que en más de la mitad de los adultos la enfermedad derivarará en forma de hemorragias intracraneales, típicamente en tálamo, ganglios basales o sustancia blanca profunda.
Cistinuria
¡¡Buenas tardes un día más!!
Hoy vamos a dedicar un ratito a hablar y comprender la cistinuria. Esta es una enfermedad que se caracteriza por la formación de unos cálculos en los uréteres, en los riñones o en la vejiga. Estos cálculos están compuestos por un aminoácido y a menudo son cálculos de cistina, tal y como se muestran en esta imagen, aunque también podrían ser de lisina, arginina u ornitina.
Esto se produce debido a que la proteína encargada de transportar estas sustancias no funciona correctamente, haciendo que se acumulen en las estructuras que acabamos de nombrar.
Hoy vamos a dedicar un ratito a hablar y comprender la cistinuria. Esta es una enfermedad que se caracteriza por la formación de unos cálculos en los uréteres, en los riñones o en la vejiga. Estos cálculos están compuestos por un aminoácido y a menudo son cálculos de cistina, tal y como se muestran en esta imagen, aunque también podrían ser de lisina, arginina u ornitina.
 |
| Fuente: www.sanar.org |
Suscribirse a:
Comentarios (Atom)